

Complejo de Dandy-Walker
Varón de 45 años, psicólogo de profesión, que consulta por ruidos en oído izquierdo desde hacía meses, pero más intenso desde hacía una semana, es como un soplo, no ha tenido mareos o vértigos, no antecedentes de trauma acústico.
Dres. Arjona Montilla C, García-Giralda M, Sánchez Rozas JA. Hospital Comarcal de Baza (Granada).
Publicación 01-07-2016
Descripción del caso clínico
- Varón de 45 años, psicólogo de profesión, que consulta por ruidos en oído izquierdo desde hacía meses, pero más intenso desde hacía una semana, es como un soplo, no ha tenido mareos o vértigos, no antecedentes de trauma acústico.
- La exploración otoscópica es normal y en la audiometría se aprecia una ligera pérdida de 30 db en ambos oídos en frecuencias agudas. Después de tratamiento con Tanakene durante 4 meses, continúa con ruidos en oído izquierdo variables, sin vértigos o cefaleas. No variación en la audiometría.
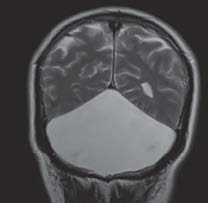
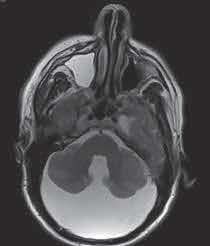
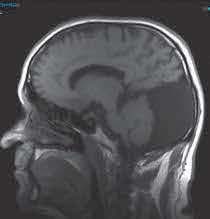
- Se solicita resonancia magnética craneal donde no se observan hallazgos de neurinoma del nervio acústico, ni en ángulo ponto-cerebeloso pero sí una gran cavidad quística en fosa posterior y de localización posterior, de 74 x 52 x 108 mm (CC x AP x T), con elevación del tentorio y desplazamiento craneal y anterior del vermis que se visualiza hipoplásico. El cuarto ventrículo se visualiza normal, existiendo una comunicación con la cavidad quística de LCR. Hallazgos compatibles con complejo de Dandy-Walker.
- Asimetría de ventrículos laterales, con un mayor aumento del VL izquierdo. No hay hidrocefalia asociada. Algún foco hiperintenso puntiforme y aislado en sustancia blanca periventricular, inespecífico y de escasa repercusión clínica.
- Cuerpo calloso, núcleos de la base, tálamos, caudados y glándula pineal, sin alteraciones significativas. Silla turca parcialmente vacía.
Discusión
El Síndrome de Dandy Walker es una anomalía congénita del cerebelo y del IV ventrículo, que aparece normalmente en la infancia. Su característica más común es la hidrocefalia y en la mayoría de los casos, las causas se desconocen.
Hay una tríada característica y que no puede faltar caracterizada por un IV ventrículo quístico, una fosa posterior agrandada y la presencia de una aplasia o hipoplasia cerebelosa; algunos han incluido otros elementos como son la atresia de los agujeros de Luschka y Magendie.
Las manifestaciones clínicas dependen de: la severidad, las malformaciones asociadas, la edad y el momento del diagnóstico. La presencia de malformaciones en el tallo cerebral puede ocasionar dificultades de succión, deglución, respiración y cardiorespiratorias. También es frecuente la hipertensión endocraneal con ataxia, espasticidad y dificultad en la motricidad fina. Suelen presentar también retraso mental.
La mayoría de los enfermos presentan síntomas durante la lactancia, con dificultades en la movilidad debido al retraso psicomotor y sin habla, el tratamiento está basado en el control de los movimientos, en la estimulación del balbuceo y en el aprendizaje de un sistema alternativo.
Es importante mencionar que se están detectando de manera incidental por motivos ajenos al síndrome de Dandy Walker, a personas asintomáticas en la edad adulta.
Aproximadamente el 68% de los enfermos presenta una o más malformaciones asociadas, que pueden estar situadas dentro y fuera del Sistema Nervioso Central. Las anomalías neurales que con mayor frecuencia se asocian al SDW son: holoprosencefalia, polimicrogiria, microcefalia, esquizencefalia, poroencefalia, agenesia del cuerpo calloso, lipoma del cuerpo calloso, estenosis del acueducto de Silvio, trastornos de la migración cerebelosos, heterotopías del núcleo olivar inferior, trastornos de la decusación del haz piramidal, encefalocele occipital, espina bífida, siringomielia, siringobulbia quiste dermoides, lipoma espinal, aneurisma de la vena magna de Galeno, lo que sugiere que las mismas son parte de las alteraciones del desarrollo general de la línea media y que ocurren en las seis primeras semanas de gestación.
Entre las no neurológicas están: angiomas faciales, labio leporino, macroglosia, paladar hendido, dismorfias faciales, hamartoma lingual, coloboma, disgenesia de retina, microftalmía, catarata, defectos septales cardíacos, persistencia del conducto arterioso, coartación de la aorta, dextrocardia, albinismo, melanosis neurocutánea, anomalías vertebrales, alteraciones cromosómicas, riñones poliquísticos, polidactilia, sindactilia.
Solo se tratan desde el punto de vista operatorio los pacientes con hidrocefalia. El abordaje quirúrgico directo del “quiste” con resección total o parcial de sus paredes o la fenestración no ofrece buenos resultados y por tanto está abandonado. Los esfuerzos se han dirigido a la solución de la hidrocefalia lo que puede obtenerse con la colocación de un sistema derivativo de LCR.
Archivo: PDF Tamaño: 0.14
Bibliografia
- Isabel Mª García Caballero. EL SÍNDROME DE DANDY WALKER Y SU INTERVENCIÓN EN LA INFANCIA. ReiDoCrea. Revista electrónica de investigación Docencia Creativa. Volumen 1. Páginas 52-58 Universidad de Granada Localizador: http:www.ugr.es/local/miguelgr/ReiDoCrea-Vol.1-Art.7-Garcia.pdf
- Goyenechea Gutiérrez, F. y Hodelín Tablada, R. Síndrome de Dandy Walker. http://www.sld.cu/galerias/pdf/sitios/neuroc/dandy_walker.pdf
- Hart, M.N., Malamud, N., Ellis, W.G. (1972). The Dandy Walker síndrome. A clinic pathological study bases on 28 cases. Neurology, 22, 771-780.
- Nazar, N. (1983). Malformación de Dandy Walker. Recuperado el 2 de mayo del 2012 de http://65.182.2.242/RMH/ pdf/1982/pdf/Vol50-3-1982-9.pdf
- López, H.J.F., García, R.R., Sánchez, V.G. y Pérez, Z.M.A. Hidrocefalia congénita asociada al Síndrome de Dandy Walker. Revisión e informe de un caso. Revista Mexicana de pediatría. http://new.medigraphic.com/cgi- bin/resumenMain.cgi?IDARTICULO=2722
- Osorio, A., Rodríguez, J.G., Pizarro, O., Koller, O., Paredes, A. y Zúñiga, L. Complejo de Dandy Walker, experiencia en el Centro de Referencia Perinatal Oriente. http://ultrasonografia.cl/us94/04OSORIODW.PDF
- Pascual-Castroviejo, I., Velez, A,, Pascual-Pascual, S.I., Roche, M.C. y Villarejo, F. (2002). Dandy-Walker malformation: analysis of 38 cases. Child’s Nervous System, 7, 88-97.
- Pascual-Castroviejo, I. (1983). Neurología Infantil. Tomo II. Barcelona: Editorial Científico Médica, 1042-1047.
- Rodríguez, J. y Cabal, A. (2009). Síndrome de Dandy Walker. Revista de AtenciónPrimaria. http://www.elsevier.es/sites/default/files/elsevier/pdf/27/27v42n01a13146044pdf
Si quieres participar enviando casos clínicos, imágenes clínicas comentadas o formación médica, puede enviárnolos a gaesmedica@cpp-proyectos.com.
PDF Tamaño: 205 kb